


Strategies for in situ reprogramming of tumor cells into antigen-presenting cells
-
摘要
免疫逃逸是肿瘤的基本特征之一,而免疫疗法近年来在肿瘤治疗中取得了巨大成功。这些疗法包括免疫检查点抑制剂、过继性细胞疗法〔如嵌合抗原受体T细胞疗法(CAR-T)和肿瘤浸润淋巴细胞(TIL)〕、癌症疫苗、溶瘤病毒等。细胞疗法在多种血液肿瘤中疗效显著,但在实体肿瘤的治疗中效果并不理想,主要原因是回输的免疫细胞难以充分浸润到肿瘤部位。为解决这一难题,研究者尝试将肿瘤组织内的癌细胞原位重编程为免疫细胞,从而充分激活抗肿瘤免疫反应并克服以往细胞治疗依赖体外细胞扩增的局限,同时具有免疫排斥反应弱的优点。目前,肿瘤原位重编程的目标细胞包括巨噬细胞、粒细胞、树突状细胞(DC)和T细胞等,其中树突状细胞作为经典抗原呈递细胞(APC)在启动免疫反应中发挥核心作用,是增强抗肿瘤免疫应答的关键效应细胞。迄今有两种原位重编程的方法,即基于转录因子异位表达及共刺激信号分子和细胞因子共表达策略,均显示出较强的转化应用前景。
AbstractImmune evasion is one of the fundamental characteristics of tumors, while immunotherapy has achieved significant success in cancer treatment in recent years. These therapies include immune checkpoint inhibitors, adoptive cell therapies [such as chimeric antigen receptor T cell therapy (CAR-T) and tumor-infiltrating lymphocytes (TIL)], cancer vaccines, and oncolytic viruses. Cell therapies have shown remarkable efficacy in various hematologic cancers, but their effect in solid tumors remains suboptimal, primarily due to the difficulty of infused immune cells in sufficiently infiltrating the tumor site. To address this challenge, researchers have explored strategies to reprogram tumor cells in situ within the tumor microenvironment into immune cells, so as to fully activate anti-tumor immune responses and overcome the limitations of conventional cell therapies that rely on in vitro cell expansion. This approach also offers the advantage of a weak immune rejection response. Currently, the target cells for in situ reprogramming include macrophages, granulocytes, dendritic cells (DCs), and T cells, among which dendritic cells, as classic antigen-presenting cells (APCs), play a central role in initiating immune responses and are key effectors in enhancing anti-tumor immunity. To date, two strategies for in situ reprogramming have been explored: one based on the ectopic expression of transcription factors and co-stimulatory molecules, and the other utilizing the co-expression of cytokines, both showing promising prospects for translational application.
-
癌症免疫治疗依赖于由肿瘤抗原特异性T淋巴细胞驱动的免疫应答的建立[1]。T细胞通过识别主要组织相容性复合体(MHC)呈递的肿瘤抗原,发挥其效应功能,包括产生炎症细胞因子和杀伤肿瘤细胞[2-3]。然而,由于肿瘤细胞的抗原呈递途径受到抑制,T细胞难以被有效激活,从而形成免疫抑制性肿瘤微环境(TME)[1]。因此,提高抗原呈递效率成为逆转免疫抑制性TME的关键之一。越来越多的研究表明,在多种癌症类型中,T细胞介导的肿瘤消退及对免疫检查点抑制剂(ICB)的反应需要经典抗原呈递细胞(APC)—尤其是1型常规树突状细胞(cDC1s)—的参与[4-7]。本文主要介绍将肿瘤细胞原位重编程为APC的两种策略,分别是基于转录因子的异位表达及共刺激信号分子和细胞因子的共表达,同时分析在不同策略中所应用的多种递送载体。通过将肿瘤细胞原位重编程为APC,可以有效激活全身抗肿瘤免疫反应并建立长期免疫,为未来广泛治疗多种肿瘤提供了可靠思路。
1. 基于转录因子异位表达的肿瘤细胞原位重编程
基于转录因子的异位表达对细胞进行重编程一直是研究的热点和难点。1987年,Davis等[8]通过过表达单个转录因子MyoD1,成功地将成纤维细胞重编程为成肌细胞。2006年,Takahashi等[9-10]通过转入外源转录因子OCT4、SOX2、KLF4和MYC成功将成纤维细胞诱导为多能干细胞(iPSC)。这一发现为后续的研究提供了新的思路,越来越多的研究人员开始探索利用不同的转录因子组合将细胞直接重编程为目的细胞。直接对细胞重编程具有特异性和高效性的优点,更适用于体内应用。然而,由于表观遗传记忆的不完全消除,直接重编程产生的细胞可能在功能和表型上不如经过iPSC阶段而分化成熟的细胞,会影响其在治疗中的应用[11]。目前,研究人员通过表达特定转录因子组合已经成功实现了多种细胞的直接重编程,并取得了良好的治疗效果。例如,通过使用腺病毒传递3种转录因子(Ngn3、Pdx1、Mafa),研究人员成功地将小鼠胰腺外分泌细胞转化为胰腺β细胞[12]。在小鼠心肌梗死模型中,利用逆转录病毒异位表达转录因子组合(Gata4、Mef2c、Tbx5),研究者将形成瘢痕的心脏成纤维细胞成功转化为心肌细胞,从而改善了心脏功能[13]。此外,在脑损伤或神经退行性疾病模型中,基于异位表达相关转录因子组合的方法也能将胶质细胞转化为功能性神经元[14]。
在肿瘤研究领域,科学家们曾尝试通过异位表达转录因子组合对肿瘤细胞直接重编程,以降低其致癌潜力,进而诱导肿瘤细胞分化[15]或转化为其他细胞类型[16-17]。然而这种方法依赖大多数肿瘤细胞的成功转化,由于重编程和传递方法的效率不足,使得这一过程面临重大挑战[18]。因此,科学家们亟需探索一种新的重编程策略,既能降低肿瘤细胞的恶性程度又能激活抗肿瘤免疫,从而在不依赖高效重编程和传递方法的情况下实现有效的抗肿瘤治疗。研究表明,将肿瘤细胞原位重编程为APC是一种理想的解决方案。
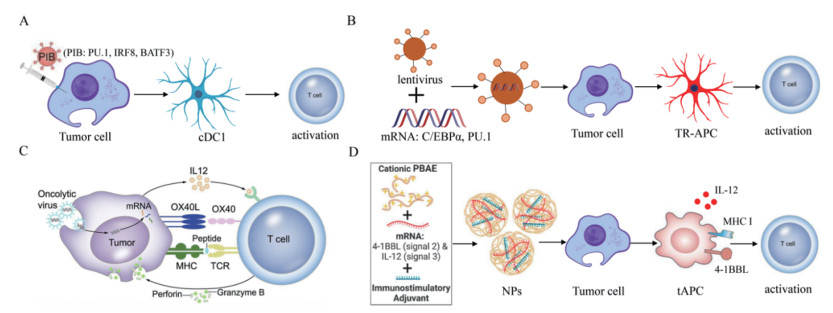
近来,有研究在肿瘤细胞重编程为具有抗原呈递功能的树突状细胞方面进行了深入而持续的探索[19]。他们通过表达转录因子PU.1、IRF8和BATF3(PIB),成功地将36例人类和小鼠血液及实体瘤细胞系重编程为1型经典树突状细胞(cDC1)(图 1A)。在重编程后的9 d内,肿瘤重编程APC(TR-APC)获得了与cDC1细胞相似的转录及表观遗传特征。此外,重编程恢复了肿瘤细胞表面抗原呈递复合物和共刺激分子的表达,使其能够在I类主要组织相容性复合体(MHC-I)上呈递内源性肿瘤抗原,并有效激活CD8+ T细胞的靶向杀伤能力。人类原代肿瘤细胞同样能够被重新编程,以增强其呈递抗原和激活患者特异性肿瘤浸润淋巴细胞的能力。以上数据表明TR-APC的产生不受细胞来源和类型的限制,适用于多种癌细胞系及原代癌细胞。除了改善抗原呈递外,TR-APC在体外和体内的致瘤性也显著降低。将体外制备的黑色素瘤衍生TR-APC注射到皮下黑色素瘤中,能够延缓肿瘤的生长并提高小鼠的存活率。此外,TR-APC引发的抗肿瘤免疫反应与免疫检查点抑制剂之间存在协同作用。
![]() 图 1 肿瘤细胞重编程为APC的主要方式A:腺病毒递送转录因子组合PIB能将肿瘤细胞原位诱导成cDC1细胞;B:通过慢病毒感染向肿瘤细胞递送转录因子C/EBPα及PU.1能使肿瘤细胞获得APC功能;C:通过溶瘤病毒向肿瘤细胞递送共刺激分子OX40L及细胞因子IL12赋予肿瘤细胞抗原呈递能力;D:借助纳米颗粒向肿瘤细胞递送共刺激分子4-1BBL及细胞因子IL12赋予肿瘤细胞抗原呈递功能。
图 1 肿瘤细胞重编程为APC的主要方式A:腺病毒递送转录因子组合PIB能将肿瘤细胞原位诱导成cDC1细胞;B:通过慢病毒感染向肿瘤细胞递送转录因子C/EBPα及PU.1能使肿瘤细胞获得APC功能;C:通过溶瘤病毒向肿瘤细胞递送共刺激分子OX40L及细胞因子IL12赋予肿瘤细胞抗原呈递能力;D:借助纳米颗粒向肿瘤细胞递送共刺激分子4-1BBL及细胞因子IL12赋予肿瘤细胞抗原呈递功能。有研究进一步开发了一种通过腺病毒递送转录因子PU.1、IRF8和BATF3的方法,在体内对肿瘤细胞进行原位重编程,使其具备类似于cDC1的抗原呈递能力,从而诱导系统性抗肿瘤免疫反应[20]。同时,研究者评估了在二维(2D)和三维(3D)环境中的肿瘤重编程的效率。结果表明,3D环境中的重编程效率与2D环境相当或更高,这表明3D环境不仅不会损害重编程,反而对其有利。在递送载体的选择方面,研究团队从穿透能力、转导效率及重编程效果等多个维度对慢病毒、腺病毒和腺相关病毒载体进行了评估,结果显示腺病毒在各个方面均表现出优越性。重编程后的肿瘤细胞能够有效重塑肿瘤微环境,招募和扩增多克隆细胞毒性T细胞,最终诱导肿瘤的消退,并在多种小鼠黑色素瘤模型中建立了长期的全身抗肿瘤免疫。
上述研究结果表明,腺病毒递送转录因子组合PIB能够将肿瘤细胞原位诱导成cDC1样细胞,从而赋予肿瘤细胞APC功能,并激活T细胞,引发系统性抗肿瘤免疫。此外,向肿瘤细胞递送其他转录因子同样可以使其获得APC功能。Linde等[21]构建了一种诱导系统,通过慢病毒感染肿瘤细胞,并使用强力霉素驱动主要髓系转录因子C/EBPα和PU.1在肿瘤细胞中的异位表达,成功地将癌细胞直接转化为TR-APC(图 1B)。进一步的研究结果显示,TR-APC不仅具备髓系细胞的表型和功能,还能够处理和呈现内源性肿瘤相关抗原,有效刺激肿瘤抗原特异性的CD4+和CD8+ T细胞。体内实验表明,TR-APC的诱导引发了癌症特异性T细胞的克隆扩增,并建立针对癌症的免疫记忆,从而促进白血病的最终治愈。此外,血液癌症和各种实体瘤,包括肉瘤和癌,都是髓系重编程为TR-APC的适用目标。原代临床标本亦可用于生成TR-APC,激活自体患者来源的T细胞,进而引发T细胞的抗肿瘤免疫反应。
2. 基于共刺激信号和细胞因子共表达的肿瘤细胞原位重编程
除了转录因子重编程外,关键共刺激信号和细胞因子的共表达也是诱导肿瘤细胞转化为APC的重要方式。溶瘤病毒是一类具复制能力的肿瘤杀伤型病毒,通过改造溶瘤病毒并感染肿瘤细胞,可以将肿瘤细胞重编程为APC。研究表明,APC激活T细胞需携带3种激活信号:信号1是通过MHC将癌症新抗原衍生的肽表位呈现给T细胞上的T细胞受体(TCR);信号2则是通过促进CD80/CD86、OX40L和(或)4-1BBL与T细胞上的受体结合提供共刺激信号;信号3是分泌细胞因子,例如白细胞介素(IL)12或IL15,以促进T细胞的存活、激活和增殖[22-23]。基于这一理论,Ye等[24]设计了一种溶瘤病毒HSV-1,以表达IL12及三聚体OX40L。在肿瘤细胞感染HSV-1并与肿瘤浸润淋巴细胞(TIL)共培养后,肿瘤细胞获得了类似APC的特性,肿瘤特异性T细胞被激活并展现出增强的肿瘤杀伤活性(图 1C)。在患者来源的异种移植物(PDX)和同基因肿瘤模型中,通过OV-OX40L/IL12与TIL的联合治疗实现了肿瘤消退。在治疗和未治疗的远端肿瘤中均取得了抗肿瘤效果,表明该联合疗法引发了针对肿瘤的全身免疫反应。此外,表现出完全反应的小鼠再移植肿瘤后肿瘤仍然无法生长,说明接受治疗的小鼠产生了抗肿瘤免疫记忆。流式细胞术分析和免疫细胞耗竭实验表明,改造后的溶瘤病毒能重塑肿瘤微环境。总体而言,OV-OX40L/IL12有效地将肿瘤细胞原位转化为APC,并增强了患者对TIL治疗的疗效。
基因递送纳米材料在再生医学的多个领域得到了广泛应用,尤其在细胞重编程方面的应用逐渐引起了研究者的关注。Tzeng等[25]设计了一种原位疫苗接种策略,该策略利用肿瘤细胞内在表达的信号1(抗原:MHC),通过安全合成的聚β-氨基酯(PBAE)基因传递纳米颗粒,直接在体内改造肿瘤细胞(图 1D)。这些纳米颗粒能够诱导共刺激分子4-1BBL的表达(信号2)并分泌细胞因子IL12(信号3)。4-1BBL已被证明可以促进CD8+ T细胞驱动的细胞毒性反应,并刺激其他免疫系统成分,包括自然杀伤(NK)细胞和APC。信号1、2和3的共表达赋予了肿瘤细胞抗原呈递的功能。在B16-F10黑色素瘤和MC38结直肠癌的小鼠模型中,重编程纳米颗粒与检查点阻断治疗相结合,显著降低了肿瘤的生长速度,甚至实现了肿瘤的清除,导致小鼠长期存活,并抑制新肿瘤的形成。体内外分析证实,局部递送的重编程纳米颗粒能够引发显著的T细胞介导的系统性细胞毒性免疫反应。
3. 总结与展望
总而言之,将肿瘤细胞原位重编程为树突状细胞是一种极具前景的免疫治疗策略。该重编程策略可分为两大类:一种是通过递送关键转录因子使肿瘤细胞转变为树突状细胞;另一种则是通过转导共刺激信号和细胞因子,促进肿瘤细胞获得抗原呈递功能。两者的根本区别在于,前者通过深度重塑细胞的转录和表观遗传状态,促使肿瘤细胞更为完全地转变为功能性树突状细胞,而后者则通过共刺激信号和细胞因子的递送,使肿瘤细胞获得抗原呈递功能。通过递送转录因子实现的重编程过程更加精准可控,但由于其引发的转化更加广泛和深入,可能伴随遗传不稳定性或其他不良反应,因此该方法的安全性值得关注。无论采用哪种策略,都需选择合适的递送载体。目前已有的载体包括慢病毒、腺病毒、腺相关病毒、溶瘤病毒和纳米递送颗粒,这些载体各具优势。腺病毒载体在转导和重编程效率上表现优异,溶瘤病毒本身可引发免疫反应,而纳米递送颗粒则相对更加安全。因此,选择适宜的递送载体至关重要,以最大程度发挥重编程效果,从而成功诱发安全的抗肿瘤免疫反应。综上所述,肿瘤细胞原位重编程为APC在肿瘤治疗领域具有广阔的应用前景,未来的研究应进一步优化基因转导方法和递送载体,以提升其临床应用的可行性和安全性。
-
[1] SHARMA P, HU-LIESKOVAN S, WARGO J A, et al. Primary, adaptive, and acquired resistance to cancer immunotherapy[J]. Cell, 2017, 168(4): 707-723. doi: 10.1016/j.cell.2017.01.017
[2] PUIG-SAUS C, SENNINO B, PENG S M, et al. Neoantigen-targeted CD8+ T cell responses with PD-1 blockade therapy[J]. Nature, 2023, 615(7953): 697-704. doi: 10.1038/s41586-023-05787-1
[3] BAWDEN E G, WAGNER T, SCHRÖDER J, et al. CD4+ T cell immunity against cutaneous melanoma encompasses multifaceted MHC II-dependent responses[J]. Science immunology, 2024, 9(91): eadi9517. doi: 10.1126/sciimmunol.adi9517
[4] SPRANGER S, DAI D, HORTON B, et al. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy[J]. Cancer cell, 2017, 31(5): 711-723. e4. doi: 10.1016/j.ccell.2017.04.003
[5] BARRY K C, HSU J, BROZ M L, et al. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments[J]. Nature medicine, 2018, 24(8): 1178-1191. doi: 10.1038/s41591-018-0085-8
[6] HUBERT M, GOBBINI E, COUILLAULT C, et al. IFN-III is selectively produced by cDC1 and predicts good clinical outcome in breast cancer[J]. Science immunology, 2020, 5(46): eaav3942. doi: 10.1126/sciimmunol.aav3942
[7] SALMON H, IDOYAGA J, RAHMAN A, et al. Expansion and activation of CD103+ dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition[J]. Immunity, 2016, 44(4): 924-938. doi: 10.1016/j.immuni.2016.03.012
[8] DAVIS R L, WEINTRAUB H, LASSAR A B. Expression of a single transfected cDNA converts fibroblasts to myoblasts[J]. Cell, 1987, 51(6): 987-1000. doi: 10.1016/0092-8674(87)90585-X
[9] TAKAHASHI K, YAMANAKA S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors[J]. Cell, 2006, 126(4): 663-676. doi: 10.1016/j.cell.2006.07.024
[10] TAKAHASHI K, TANABE K, OHNUKI M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors[J]. Cell, 2007, 131(5): 861-872. doi: 10.1016/j.cell.2007.11.019
[11] CAHAN P, LI H, MORRIS S A, et al. CellNet: network biology applied to stem cell engineering[J]. Cell, 2014, 158(4): 903-915. doi: 10.1016/j.cell.2014.07.020
[12] ZHOU Q, BROWN J, KANAREK A, et al. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells [J]. Nature, 2008, 455(7213): 627-632. doi: 10.1038/nature07314
[13] QIAN L, HUANG Y, SPENCER C I, et al. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes[J]. Nature, 2012, 485(7400): 593-598. doi: 10.1038/nature11044
[14] TORPER O, OTTOSSON D R, PEREIRA M, et al. In vivo reprogramming of striatal NG2 Glia into functional neurons that integrate into local host circuitry[J]. Cell reports, 2015, 12(3): 474-481. doi: 10.1016/j.celrep.2015.06.040
[15] PARK N I, GUILHAMON P, DESAI K, et al. ASCL1 reorganizes chromatin to direct neuronal fate and suppress tumorigenicity of glioblastoma stem cells[J]. Cell stem cell, 2017, 21(3): 411. doi: 10.1016/j.stem.2017.08.008
[16] RAPINO F, ROBLES E F, RICHTER-LARREA J A, et al. C/EBPα induces highly efficient macrophage transdifferentiation of B lymphoma and leukemia cell lines and impairs their tumorigenicity[J]. Cell reports, 2017, 19(6): 1281. doi: 10.1016/j.celrep.2017.04.072
[17] MCCLELLAN J S, DOVE C, GENTLES A J, et al. Reprogramming of primary human Philadelphia chromosome-positive B cell acute lymphoblastic leukemia cells into nonleukemic macrophages[J]. Proceedings of the national academy of sciences of the United States of America, 2015, 112(13): 4074-4079.
[18] WANG H F, YANG Y C, LIU J D, et al. Direct cell reprogramming: approaches, mechanisms and progress[J]. Nature reviews molecular cell biology, 2021, 22(6): 410-424. doi: 10.1038/s41580-021-00335-z
[19] ZIMMERMANNOVA O, FERREIRA A G, ASCIC E, et al. Restoring tumor immunogenicity with dendritic cell reprogramming[J]. Science immunology, 2023, 8(85): eadd4817. doi: 10.1126/sciimmunol.add4817
[20] ASCIC E, ÅKERSTRÖM F, SREEKUMAR NAIR M, et al. In vivo dendritic cell reprogramming for cancer immunotherapy[J]. Science, 2024, 386(6719): eadn9083. doi: 10.1126/science.adn9083
[21] LINDE M H, FAN A C, KÖHNKE T, et al. Reprogramming cancer into antigen-presenting cells as a novel immunotherapy[J]. Cancer discovery, 2023, 13(5): 1164-1185. doi: 10.1158/2159-8290.CD-21-0502
[22] CHARETTE M D, MARABELLE A, HOUOT R. Turning tumour cells into antigen presenting cells: the next step to improve cancer immunotherapy? [J]. European journal of cancer, 2016, 68: 134-147. doi: 10.1016/j.ejca.2016.09.010
[23] ETXEBERRIA I, OLIVERA I, BOLAÑOS E, et al. Engineering bionic T cells: signal 1, signal 2, signal 3, reprogramming and the removal of inhibitory mechanisms[J]. Cellular & molecular immunology, 2020, 17(6): 576-586.
[24] YE K, LI F, WANG R K, et al. An armed oncolytic virus enhances the efficacy of tumor-infiltrating lymphocyte therapy by converting tumors to artificial antigen-presenting cells in situ[J]. Molecular therapy, 2022, 30(12): 3658-3676. doi: 10.1016/j.ymthe.2022.06.010
[25] TZENG S Y, PATEL K K, WILSON D R, et al. In situ genetic engineering of tumors for long-lasting and systemic immunotherapy[J]. Proceedings of the national academy of sciences of the United States of America, 2020, 117(8): 4043-4052.
 下载:
下载:
计量
- 文章访问数: 37
- HTML全文浏览量: 8
- PDF下载量: 9
